OmicsFlow
🧬 OmicsFlow
A modular, containerized NGS pipeline for RNA-seq, long-read, and metagenomic analysis
![]()
![]()
![]() —
—
📋 Overview
OmicsFlow is a production-ready bioinformatics pipeline built with Nextflow and Docker, designed for reproducible multi-omics data analysis. It supports three major sequencing technologies and workflows:
| Workflow | Technology | Key tools | Status |
|---|---|---|---|
rnaseq.nf |
Illumina short reads | FastQC · STAR · Salmon · DESeq2 | ✅ Stable |
longread.nf |
Oxford Nanopore (ONT) | NanoStat · Minimap2 · Samtools | 🚧 In development |
metagenomics.nf |
Illumina / ONT | Kraken2 · Bracken | 🚧 In development |
All workflows are fully containerized via Docker and can run locally, on HPC clusters (SLURM/PBS), or in the cloud (AWS Batch).
📸 Pipeline Results
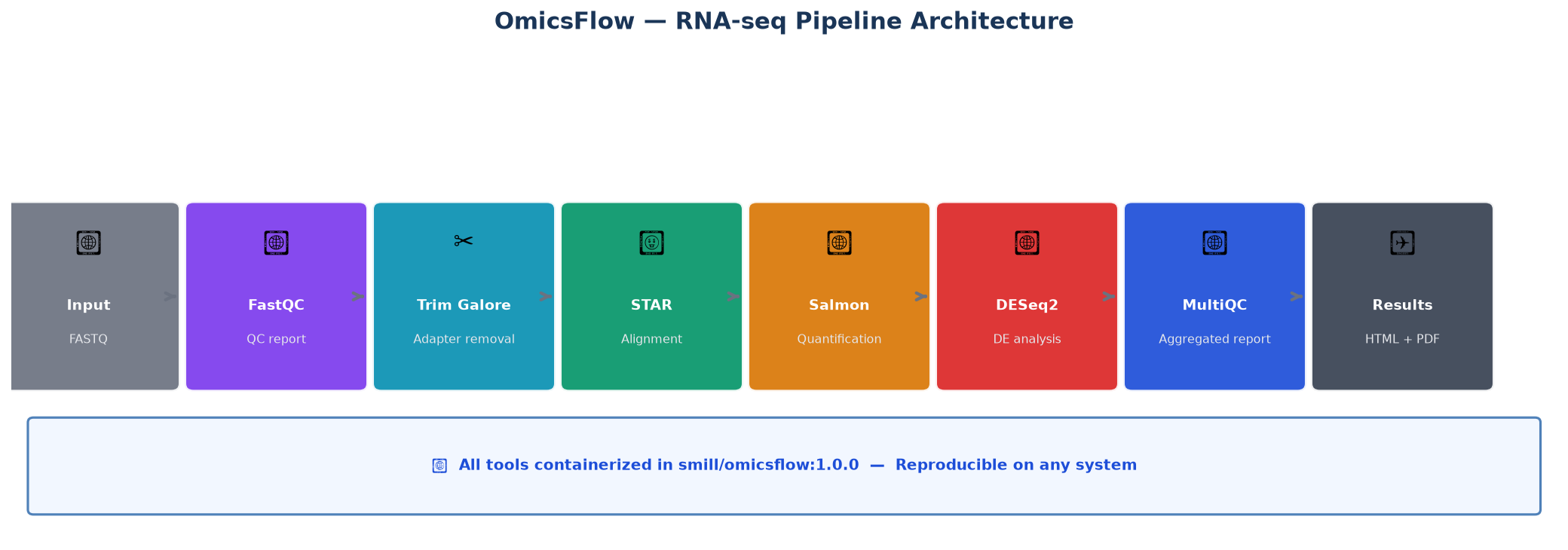
Pipeline Architecture
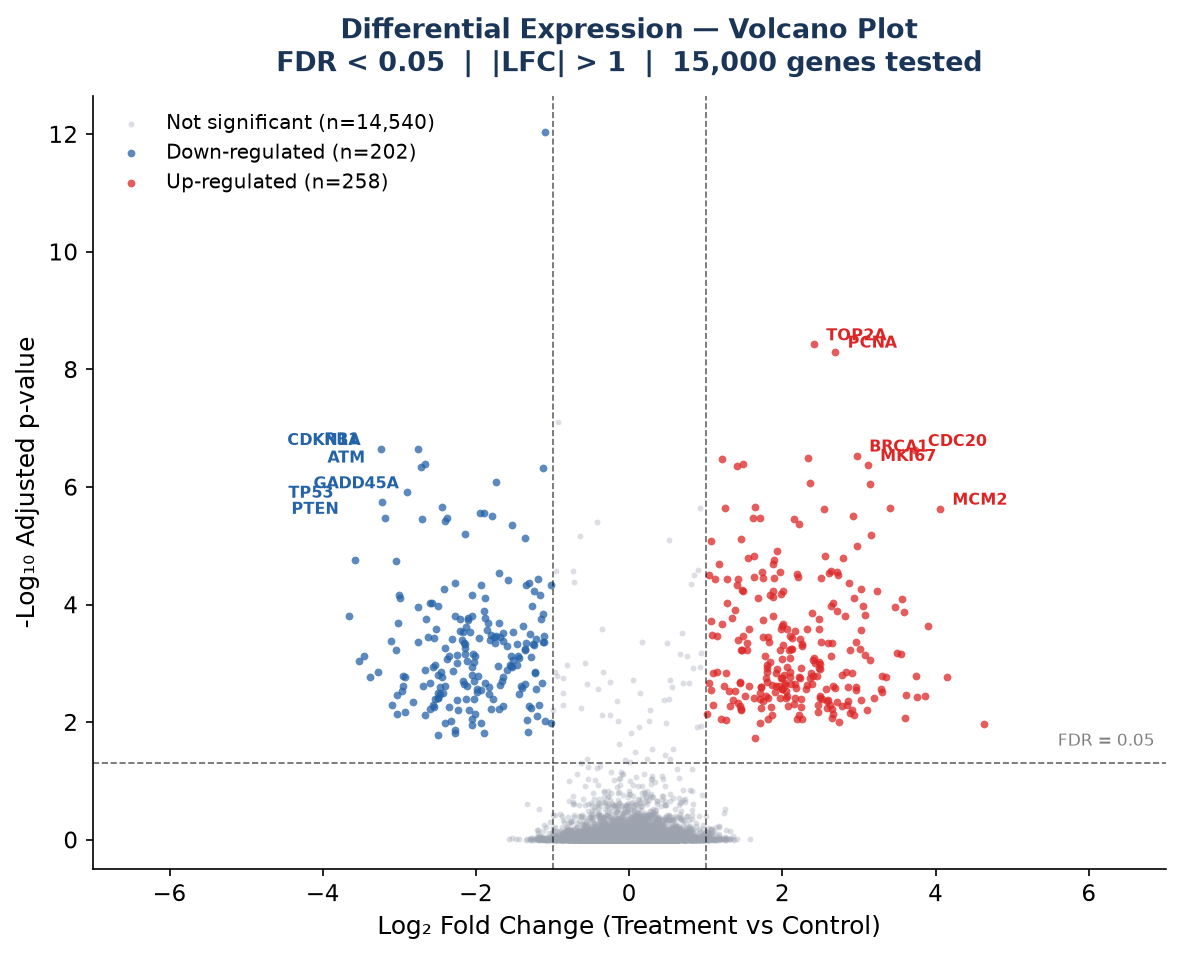
Differential Expression — Volcano Plot
Top 50 DE Genes — Heatmap

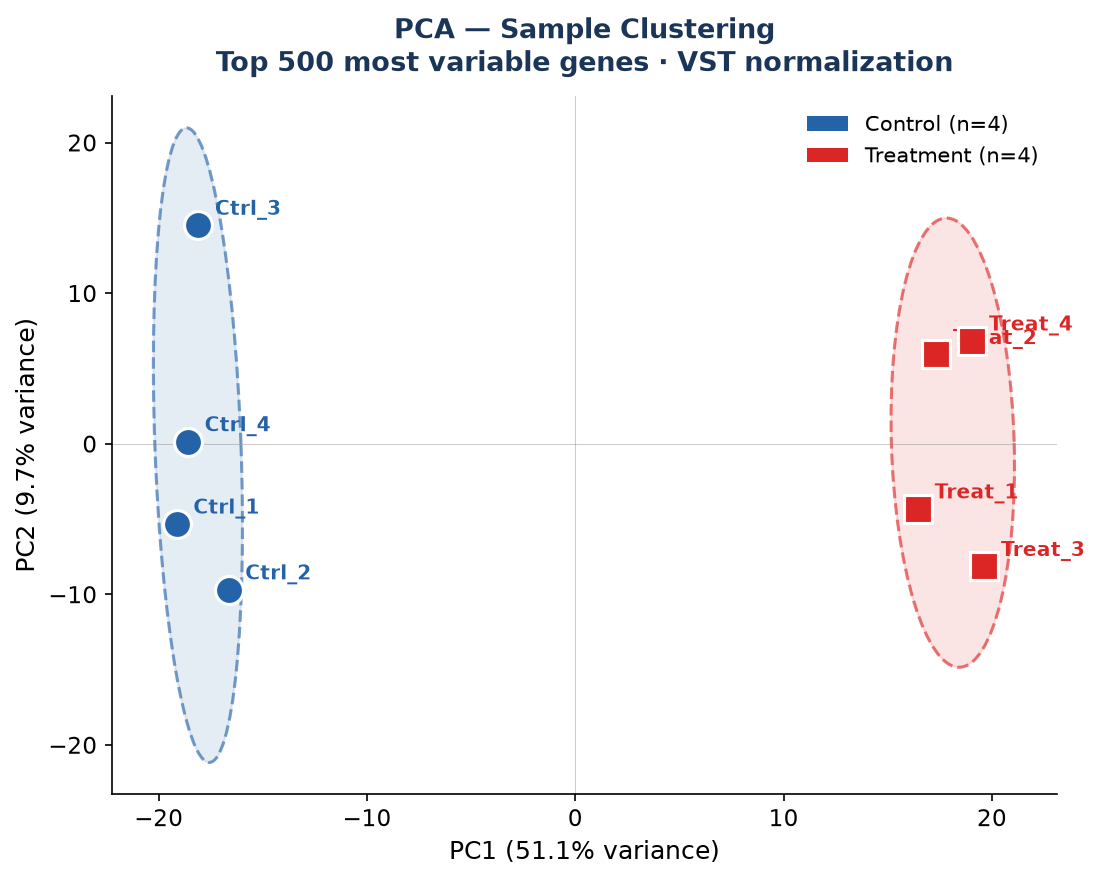
Sample Clustering — PCA
📊 Validation Metrics
Benchmarked on nf-core test dataset (S. cerevisiae, GSE110004, 4 samples × 50,000 reads):
| Metric | Value |
|---|---|
| Input reads per sample | 50,000 |
| Reads passing QC | 99.5% |
| Adapter contamination (auto-detected & removed) | 40.3% |
| Uniquely mapped reads (STAR) | 81.8% – 84.6% |
| Properly paired reads | 100% |
| Mismatch rate | 0.9% |
| Pipeline execution time (4 samples, 4 CPUs) | ~8 min |
| Docker image size | 4.63 GB |
📦 What you need before starting
OmicsFlow is flexible — you can use the full pipeline or individual tools depending on your needs.
The only real requirement: your data
| Use case | What you need |
|---|---|
| Quality control only | FASTQ files |
| Trimming only | FASTQ files |
| Alignment (STAR) | FASTQ files + reference genome + GTF + STAR index |
| Quantification (Salmon) | FASTQ files + Salmon index |
| Statistics (Samtools) | An existing BAM file |
| Differential expression | Salmon counts + sample metadata |
| Python / R analysis | Your own data + scripts |
You do not need to prepare everything upfront. Start with what you have and add steps as needed.
Reference genome & annotation (only if using STAR alignment)
If you plan to use STAR for alignment, you need a reference genome and its annotation.
If you already have a STAR index on your server or HPC — just point to it with --genomeDir. No need to rebuild it. Any STAR-compatible index works, regardless of how it was generated.
If you need to build one (one-time operation, ~45 min for full human genome):
# Download reference genome (human GRCh38)
wget https://ftp.ensembl.org/pub/release-109/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
# Download gene annotation
wget https://ftp.ensembl.org/pub/release-109/gtf/homo_sapiens/Homo_sapiens.GRCh38.109.gtf.gz
gunzip Homo_sapiens.GRCh38.109.gtf.gz
# Build STAR index using OmicsFlow Docker
docker run --rm -v $(pwd):/data smill/omicsflow:1.0.0 \
bash -c "mkdir -p /data/star_index && STAR --runMode genomeGenerate \
--genomeDir /data/star_index \
--genomeFastaFiles /data/genome/GRCh38.fa \
--sjdbGTFfile /data/genome/GRCh38.gtf \
--runThreadN 8"
⚠️ Build the index once, store it, reuse it forever for all your experiments.
What you do NOT need to install
Everything is already inside the Docker image:
| Tool | Without OmicsFlow | With OmicsFlow |
|---|---|---|
| FastQC | Manual install | ✅ Included |
| Trim Galore | Manual install | ✅ Included |
| STAR | Compile from source | ✅ Included |
| Salmon | Manual install | ✅ Included |
| Samtools | Compile from source | ✅ Included |
| DESeq2 | R + Bioconductor setup | ✅ Included |
| MultiQC | pip install | ✅ Included |
| BioPython | pip install | ✅ Included |
| numpy / pandas / matplotlib | pip install | ✅ Included |
| NanoStat / NanoPlot | pip install | ✅ Included |
| Kraken2 | Manual install | ✅ Included |
| Minimap2 | Manual install | ✅ Included |
🐳 Run with Docker only (no Nextflow required)
The easiest way to use OmicsFlow — just Docker, no installation needed.
# Pull the image
docker pull smill/omicsflow:1.0.0
# Step 1 — Quality control (FastQC)
docker run --rm -v $(pwd)/data:/data smill/omicsflow:1.0.0 \
bash -c "fastqc /data/sample_R1.fastq.gz /data/sample_R2.fastq.gz --outdir /data/qc"
# Step 2 — Adapter trimming (Trim Galore)
docker run --rm -v $(pwd)/data:/data smill/omicsflow:1.0.0 \
bash -c "trim_galore --paired --cores 4 \
/data/sample_R1.fastq.gz /data/sample_R2.fastq.gz \
-o /data/trimmed"
# Step 3 — Alignment (STAR)
docker run --rm -v $(pwd)/data:/data smill/omicsflow:1.0.0 \
bash -c "STAR --runMode alignReads \
--genomeDir /data/star_index \
--readFilesIn /data/trimmed/sample_R1_val_1.fq.gz /data/trimmed/sample_R2_val_2.fq.gz \
--readFilesCommand zcat \
--outSAMtype BAM SortedByCoordinate \
--outFileNamePrefix /data/aligned/sample. \
--runThreadN 4"
# Step 4 — Quantification (Salmon)
docker run --rm -v $(pwd)/data:/data smill/omicsflow:1.0.0 \
bash -c "salmon quant \
--index /data/salmon_index \
--libType A \
-1 /data/trimmed/sample_R1_val_1.fq.gz \
-2 /data/trimmed/sample_R2_val_2.fq.gz \
--output /data/counts/sample \
--threads 4 \
--validateMappings"
# Step 5 — BAM statistics (Samtools)
docker run --rm -v $(pwd)/data:/data smill/omicsflow:1.0.0 \
bash -c "samtools flagstat /data/aligned/sample.Aligned.sortedByCoord.out.bam"
# Step 6 — Aggregated QC report (MultiQC)
docker run --rm -v $(pwd)/data:/data smill/omicsflow:1.0.0 \
bash -c "multiqc /data --outdir /data/multiqc"
# Interactive R session (DESeq2, ggplot2...)
docker run --rm -it -v $(pwd)/data:/data smill/omicsflow:1.0.0 R
# Interactive Python session (biopython, pandas, matplotlib...)
docker run --rm -it -v $(pwd)/data:/data smill/omicsflow:1.0.0 python3
Windows users: replace
$(pwd)with%cd%in CMD, or use the full path.
🚀 Run with Nextflow (recommended for production)
Prerequisites
- Nextflow ≥ 22.10
- Docker or Singularity
- Java 17+
Run in one command
# Clone the repository
git clone https://github.com/Millimono/OmicsFlow.git
cd OmicsFlow
# Run RNA-seq pipeline with test data
nextflow run workflows/rnaseq.nf \
--input data/test/samplesheet.csv \
--genome GRCh38 \
--outdir results/ \
-profile docker
Input samplesheet format (CSV)
sample,fastq_1,fastq_2,strandedness
ctrl_rep1,/path/to/ctrl_rep1_R1.fastq.gz,/path/to/ctrl_rep1_R2.fastq.gz,reverse
ctrl_rep2,/path/to/ctrl_rep2_R1.fastq.gz,/path/to/ctrl_rep2_R2.fastq.gz,reverse
treat_rep1,/path/to/treat_rep1_R1.fastq.gz,/path/to/treat_rep1_R2.fastq.gz,reverse
treat_rep2,/path/to/treat_rep2_R1.fastq.gz,/path/to/treat_rep2_R2.fastq.gz,reverse
Strandedness: use
reversefor most Illumina TruSeq kits,forwardfor some stranded protocols,unstrandedif unsure.
🗂️ Project Structure
OmicsFlow/
├── workflows/
│ ├── rnaseq.nf # ✅ RNA-seq Illumina pipeline (stable)
│ ├── longread.nf # 🚧 Nanopore long-read pipeline (in development)
│ └── metagenomics.nf # 🚧 Metagenomic pipeline (in development)
│
├── modules/
│ ├── qc/ # FastQC, MultiQC, NanoStat
│ ├── alignment/ # STAR, Minimap2, Samtools
│ └── quantification/ # Salmon, DESeq2
│
├── analysis/
│ ├── deseq2.R # Differential expression (DESeq2 / edgeR)
│ ├── plots.py # Heatmaps, volcano plots, PCA
│ └── report.Rmd # Automated HTML report template
│
├── containers/
│ └── Dockerfile # All tools in one reproducible image
│
├── data/
│ └── test/ # Public mini-datasets for testing
│ ├── samplesheet.csv
│ └── reads/ # nf-core GSE110004 subset (Illumina)
│
├── docs/ # Documentation (GitHub Pages)
├── .github/
│ └── workflows/
│ └── ci.yml # GitHub Actions CI/CD
└── nextflow.config # Profiles: local, cluster, cloud
📊 Workflows in Detail
1. RNA-seq Pipeline (rnaseq.nf) ✅ Stable
Designed for bulk RNA-seq analysis from raw FASTQ to differential expression.
Input FASTQ
│
▼
[FastQC] ──────────────────────────> QC report
│
▼
[Trim Galore] ──> Trimmed reads
│
▼
[STAR] ──> Aligned BAM + splice junctions
│
▼
[Salmon] ──> Gene/transcript counts
│
▼
[DESeq2 / edgeR] ──> Differential expression
│
▼
[MultiQC] ──> Aggregated QC report (HTML)
Output files:
results/qc/— FastQC + MultiQC reportsresults/aligned/— BAM files + indexresults/counts/— Salmon quantificationresults/deseq2/— DE results, volcano plots, heatmapsresults/report.html— Full automated HTML report
2. Long-read Pipeline (longread.nf) 🚧 In development
For Nanopore sequencing data. Coming soon — tools already available in the Docker image.
Input FASTQ (ONT)
│
▼
[NanoStat / NanoPlot] ──> Read quality stats
│
▼
[Minimap2] ──> Aligned BAM
│
▼
[Samtools] ──> Sorted + indexed BAM
│
▼
[MultiQC] ──> Aggregated report
In the meantime, you can use these tools individually via Docker — see the Docker section above.
3. Metagenomic Pipeline (metagenomics.nf) 🚧 In development
Taxonomic classification and abundance profiling. Coming soon — Kraken2 already available in the Docker image.
Input FASTQ
│
▼
[FastQC + Trim Galore] ──> Clean reads
│
▼
[Kraken2] ──> Taxonomic classification
│
▼
[Bracken] ──> Abundance re-estimation
In the meantime, you can run Kraken2 directly:
docker run --rm -v $(pwd):/data smill/omicsflow:1.0.0 \ bash -c "kraken2 --db /data/kraken2_db --paired \ /data/R1.fastq.gz /data/R2.fastq.gz \ --output /data/kraken2_output.txt \ --report /data/kraken2_report.txt"
🛠️ Technical Stack
| Category | Tools | Versions |
|---|---|---|
| Pipeline orchestration | Nextflow DSL2 | ≥ 22.10 |
| Containerization | Docker · Singularity | 28.x |
| QC | FastQC · MultiQC · NanoStat · NanoPlot | 0.12.1 · 1.35 · 1.6.0 |
| Alignment | STAR · Minimap2 | 2.7.11b · 2.31 |
| Quantification | Salmon | 1.12.0 |
| Variant calling | Samtools · BCFtools | 1.23.1 |
| Metagenomics | Kraken2 | 2.1.3 |
| Statistical analysis | DESeq2 · edgeR · R | R 4.5.2 |
| Visualization | ggplot2 · matplotlib · seaborn | — |
| Languages | Python · R · Bash · C · C++ | Python 3.x |
| CI/CD | GitHub Actions | — |
| Documentation | GitHub Pages | — |
🧪 Test Data
Test data used during development (publicly available):
| Dataset | Source | Size | Used for |
|---|---|---|---|
| GSE110004 / SRR6357070-71 (4 samples) | nf-core test datasets | ~8 MB | RNA-seq validation |
| S. cerevisiae R64-1-1 genome | nf-core test datasets | ~230 KB | Reference genome |
| S. cerevisiae gene annotation | nf-core test datasets | ~200 KB | Gene annotation |
⚙️ Configuration
OmicsFlow supports multiple execution profiles defined in nextflow.config:
profiles {
docker {
docker.enabled = true
process.executor = 'local'
}
cluster {
process.executor = 'slurm'
singularity.enabled = true
process.queue = 'normal'
}
cloud {
process.executor = 'awsbatch'
aws.region = 'ca-central-1'
}
test {
params.input = "${projectDir}/data/test/samplesheet.csv"
params.outdir = 'results_test'
docker.enabled = true
}
}
📈 Results & Outputs
Every run generates a timestamped output directory:
results/
├── qc/
│ ├── fastqc/ # Per-sample FastQC reports (HTML)
│ └── multiqc_report.html # Aggregated QC report
├── trimmed/
│ └── logs/ # Trim Galore trimming reports
├── aligned/
│ ├── sample.Aligned.sortedByCoord.out.bam
│ └── sample.Log.final.out # Mapping statistics
├── counts/
│ └── salmon/ # Transcript-level quantification
│ └── quant.sf
├── deseq2/
│ ├── deseq2_results.csv # DE genes table
│ ├── volcano_plot.pdf # Volcano plot
│ ├── heatmap_top50.pdf # Top 50 DE genes heatmap
│ └── pca_plot.pdf # PCA plot
└── pipeline_info/
├── execution_report.html
└── execution_timeline.html
🔗 Link to Research
This pipeline was developed in conjunction with research in AI-based medical imaging and bioinformatics:
- MalariaScan — AI detection of malaria via blood microscopy. Prix Coup de cœur Jean-Marc Léger, UdeM 2025.
- HAtt-CNN — Adaptive visual attention supervision with heuristic masks for CNN interpretability. (Under review 2026)
- EpitopeNet — Backpropagation-free prototype learning inspired by B-cell dynamics for mammography classification. 76.03% accuracy on MiniDDSM. (Under review 2026)
The
analysis/module is extensible — ML models from the above projects can be integrated as additional pipeline steps.
📚 Documentation
Full documentation available at: millimono.github.io/OmicsFlow
🤝 Contributing
Contributions welcome! Please open a pull request.
git clone https://github.com/Millimono/OmicsFlow.git
cd OmicsFlow
git checkout -b feature/my-new-module
📄 Citation
Millimono, S. (2026). OmicsFlow: A modular containerized NGS pipeline
for reproducible multi-omics analysis (v1.0.1). Zenodo.
https://doi.org/10.5281/zenodo.20677900
👤 Author
Sory Millimono PhD Candidate in AI · Bioinformatician Université de Montréal & Mohammed V University – ENSIAS
- 📧 millimono64.sm@gmail.com
- 🎓 Google Scholar — h-index 1 · 24 citations
- 🔬 ORCID: 0009-0005-1960-9136
📜 License
MIT License — see LICENSE for details.